First-line Laboratory tests
| Test | APTT(1) (activated cephalin time) |
Mixing test(2) | PT (prothrombin time) |
Fibrinogen | TT (thrombin time) |
BT(3) (bleeding time) |
Platelet count(4) |
|---|---|---|---|---|---|---|---|
| Result | Increased | Corrected | Normal | Normal | Normal | Normal | Normal |
(1) APTT: "inconsistent" prolongation of APTT occurs; lengthening depends on levels of factor VIII present and on the sensitivity to factor VIII of the reagent being used.
(2) Mixing test: Prolongation of APTT is corrected by mixing the patient's plasma in equal parts with a pool of normal plasma.
(3) BT: bleeding time (Ivy method) is only increased in around 50% of cases; it is never increased in type 2N von Willebrand disease; in vitro tests to reproduce bleeding time can provide diagnosis (except in type 2N von Willebrand disease).
(4) Platelet count: this parameter is normal in most forms of von Willebrand disease. The platelet count may be depressed in some patients with type 2B von Willebrand disease, in whom fluctuating thrombocytopenia of varying severity may be seen.
Specialized tests
VWF ristocetin cofactor activity
- Assay of von Willebrand factor ristocetin cofactor activity measures ristocetin-induced binding of von Willebrand factor to platelet glycoprotein GPIb. This activity is decreased in all types of von Willebrand disease except type 2N.Von
Willebrand factor antigen
• Assay of von Willebrand factor antigen allows differential diagnosis between quantitative deficits, in which it is decreased, and qualitative deficits, in which it is normal and in which the ratio VWF:RCo / VWF Ag is < 0.7.
As a general rule, von Willebrand factor levels vary according to blood group and are lower in people with blood type O than in non-O subjects.
Binding to factor VIII
• Assay of factor VIII (FVIII:C) generally reveals a decrease, although it is still higher than the level of von Willebrand factor, except in type 2N von Willebrand disease.Determination of the binding capacity of factor VIII for von Willebrand factor allows differential diagnosis with regard to haemophilia A.
Collagen binding assay
• Assay of collagen binding of von Willebrand factor (VWF:CB) can be used to detect abnormalities in VWF multimerisation.
Differential diagnosis
Haemophilia A
Factor VIII is reduced whereas von Willebrand factor ristocetin cofactor is normal, as is binding of factor VIII to von Willebrand factor.
Pseudo (platelet type) von Willebrand disease
Pseudo von Willebrand disease is a platelet disease involving increased affinity of GPIb platelets for von Willebrand factor with variable levels of thrombocytopenia. Differential diagnosis between pseudo von Willebrand disease and type 2B von Willebrand disease can only be performed at specialised laboratories.
Acquired von Willebrand disease
Acquired VWF deficiency can be seen in certain clinical settings such as lymphoproliferative and myeloproliferative syndromes, certain types of cancer and certain autoimmune diseases, as well as in aortic valve stenosis.
This acquired deficiency may be related to the presence of auto-antibodies directed against VWF, to the adsorption of VWF at the surface of certain cells, or to degradation of VWF.
- Symptoms are generally moderate.
- Onset normally occurs after the age of 50 years.
- Laboratory signs are identical to those of von Willebrand disease.
- Disappearance of the disease following aetiological treatment may serve as a basis for retrospective diagnosis.
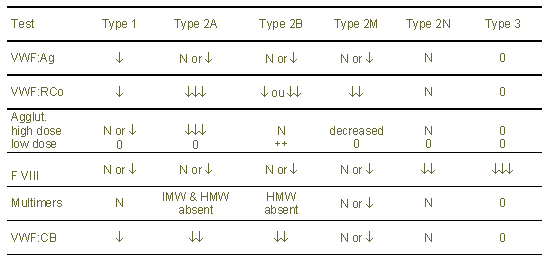
Table 1: Different types of von Willebrand disease and associated tests